
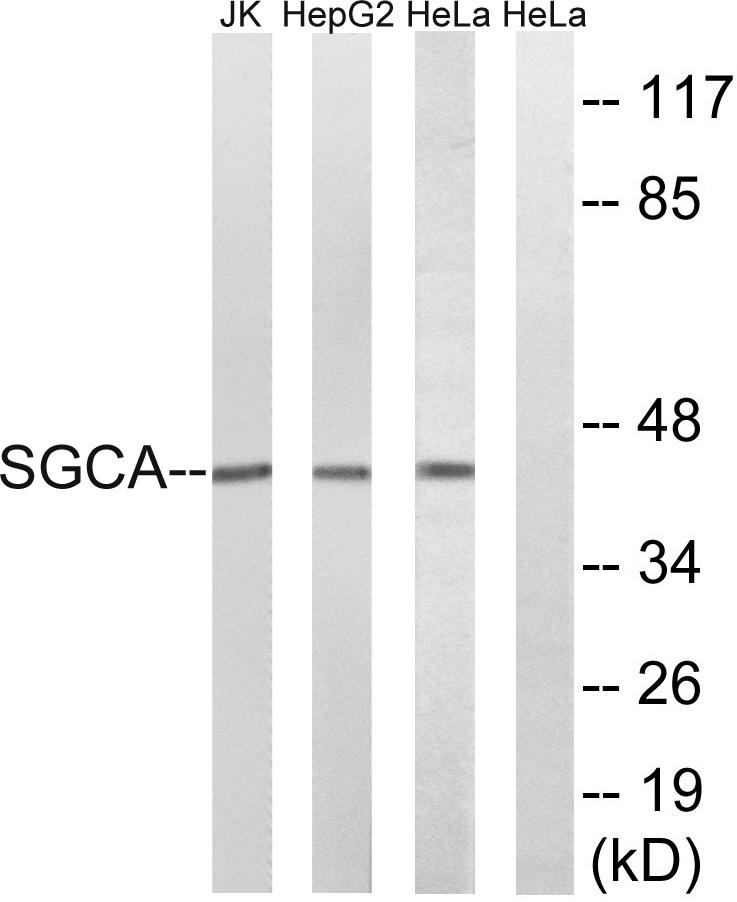

Target:Sarcoglycan α
Fields:Hypertrophic cardiomyopathy;Arrhythmogenic right ventricular cardiomyopathy;Dilated cardiomyopathy;Viral myocarditis
Gene Name:SGCA
Protein Name:Alpha-sarcoglycan
Human Gene Id:6442
Human Swiss Prot No:Q16586
Mouse Gene Id:20391
Mouse Swiss Prot No:P82350
Immunogen:The antiserum was produced against synthesized peptide derived from human SGCA. AA range:161-210
Specificity:Sarcoglycan α Polyclonal Antibody detects endogenous levels of Sarcoglycan α protein.
Formulation:Liquid in PBS containing 50% glycerol, 0.5% BSA and 0.02% sodium azide.
Source:Polyclonal, Rabbit,IgG
Dilution:WB 1:500-2000;IHC 1:50-300
Purification:The antibody was affinity-purified from rabbit antiserum by affinity-chromatography using epitope-specific immunogen.
Concentration:1 mg/ml
Storage Stability:-15°C to -25°C/1 year(Do not lower than -25°C)
Other Name:SGCA;ADL;DAG2;Alpha-sarcoglycan;Alpha-SG;50 kDa dystrophin-associated glycoprotein;50DAG;Adhalin;Dystroglycan-2
Observed Band(KD):43kD
Background:sarcoglycan alpha(SGCA) Homo sapiens This gene encodes a component of the dystrophin-glycoprotein complex (DGC), which is critical to the stability of muscle fiber membranes and to the linking of the actin cytoskeleton to the extracellular matrix. Its expression is thought to be restricted to striated muscle. Mutations in this gene result in type 2D autosomal recessive limb-girdle muscular dystrophy. Multiple transcript variants encoding different isoforms have been found for this gene. [provided by RefSeq, Oct 2008],
Function:disease:Defects in SGCA are the cause of limb-girdle muscular dystrophy type 2D (LGMD2D) [MIM:608099]; also known as Duchenne-like muscular dystrophy autosomal recessive type 2 or severe childhood autosomal recessive muscular dystrophy (SCARMD). LGMD2D is an autosomal recessive degenerative myopathy characterized by progressive muscle wasting from early childhood with loss of independent ambulation by teenage years. Muscle biopsy shows necrosis, decreased immunostaining for alpha sarcoglycan, and adhalin deficiency. The phenotype is less severe than LGMD2C.,function:Component of the sarcoglycan complex, a subcomplex of the dystrophin-glycoprotein complex which forms a link between the F-actin cytoskeleton and the extracellular matrix.,online information:SGCA mutations in LGMD2D,similarity:Belongs to the sarcoglycan alpha/epsilon family.,subunit:Interacts with the syntrophin SNTA1. Cross-
Subcellular Location:Cell membrane, sarcolemma ; Single-pass type I membrane protein . Cytoplasm, cytoskeleton .
Expression:Most strongly expressed in skeletal muscle. Also expressed in cardiac muscle and, at much lower levels, in lung. In the fetus, most abundant in cardiac muscle and, at lower levels, in lung. Also detected in liver and kidney. Not expressed in brain.
商品信息已成功复制,启研竭诚为您服务




