

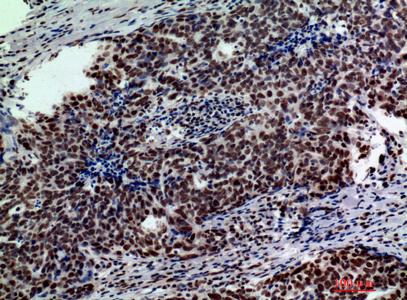
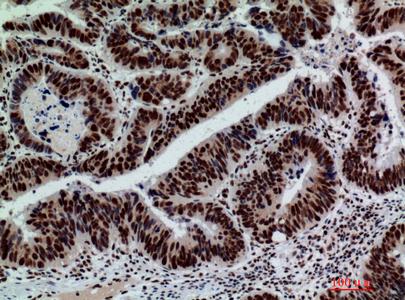
Target:Galactosidase β
Fields:Galactose metabolism;Other glycan degradation;Glycosaminoglycan degradation;Sphingolipid metabolism;Glycosphingolipid biosynthesis - ganglio series;Metabolic pathways;Lysosome
Gene Name:GLB1
Protein Name:Beta-galactosidase
Human Gene Id:2720
Human Swiss Prot No:P16278
Mouse Swiss Prot No:P23780
Immunogen:Synthesized peptide derived from the Internal region of human Galactosidase β.
Specificity:Galactosidase β Polyclonal Antibody detects endogenous levels of Galactosidase β protein.
Formulation:Liquid in PBS containing 50% glycerol, 0.5% BSA and 0.02% sodium azide.
Source:Polyclonal, Rabbit,IgG
Dilution:WB 1:500 - 1:2000. IHC: 1:100-1:300. ELISA: 1:10000.. IF 1:50-200
Purification:The antibody was affinity-purified from rabbit antiserum by affinity-chromatography using epitope-specific immunogen.
Concentration:1 mg/ml
Storage Stability:-15°C to -25°C/1 year(Do not lower than -25°C)
Other Name:GLB1;ELNR1;Beta-galactosidase;Acid beta-galactosidase;Lactase;Elastin receptor 1
Observed Band(KD):76kD
Background: This gene encodes a member of the glycosyl hydrolase 35 family of proteins. Alternative splicing results in multiple transcript variants, at least one of which encodes a preproprotein that is proteolytically processed to generate the mature lysosomal enzyme. This enzyme catalyzes the hydrolysis of a terminal beta-linked galactose residue from ganglioside substrates and other glycoconjugates. Mutations in this gene may result in GM1-gangliosidosis and Morquio B syndrome. [provided by RefSeq, Nov 2015],
Function:catalytic activity:Hydrolysis of terminal non-reducing beta-D-galactose residues in beta-D-galactosides.,disease:Defects in GLB1 are the cause of GM1-gangliosidosis type 1 (GM1G1) [MIM:230500]; also known as infantile GM1-gangliosidosis. GM1-gangliosidosis is an autosomal recessive lysosomal storage disease marked by the accumulation of GM1 gangliosides, glycoproteins and keratan sulfate primarily in neurons of the central nervous system. GM1G1 is characterized by onset within the first three months of life, central nervous system degeneration, coarse facial features, hepatosplenomegaly, skeletal dysmorphology reminiscent of Hurler syndrome, and rapidly progressive psychomotor deterioration. Urinary oligosaccharide levels are high. It leads to death usually between the first and second year of life.,disease:Defects in GLB1 are the cause of GM1-gangliosidosis type 2 (GM1G2) [MIM:230600];
Subcellular Location:[Isoform 1]: Lysosome .; [Isoform 2]: Cytoplasm, perinuclear region . Localized to the perinuclear area of the cytoplasm but not to lysosomes. .
Expression:Detected in placenta (at protein level) (PubMed:8383699). Detected in fibroblasts and testis (PubMed:2511208).
商品信息已成功复制,启研竭诚为您服务





