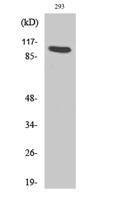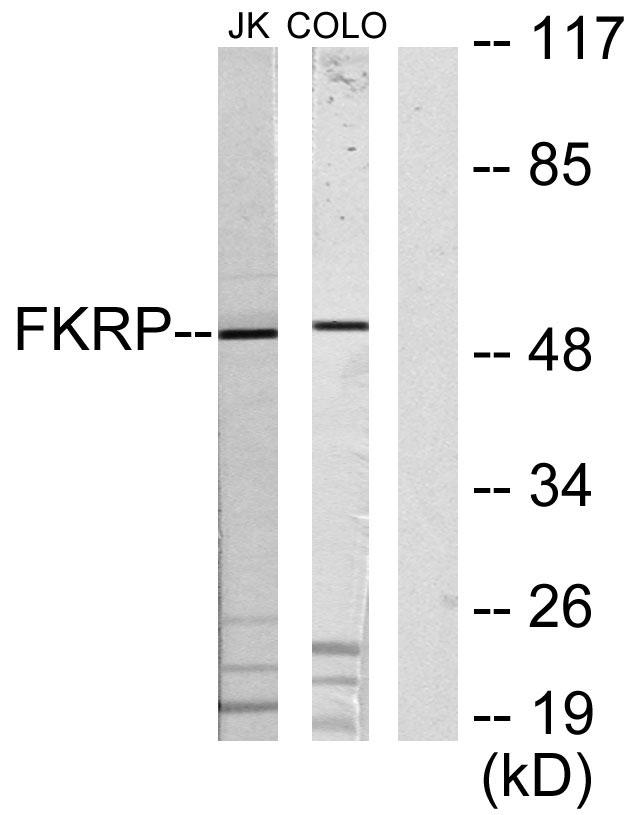
Target:FKRP
Fields:Mannose type O-glycan biosynthesis;Metabolic pathways
Gene Name:FKRP
Protein Name:Fukutin-related protein
Human Gene Id:79147
Human Swiss Prot No:Q9H9S5
Mouse Gene Id:243853
Mouse Swiss Prot No:Q8CG64
Immunogen:The antiserum was produced against synthesized peptide derived from human FKRP. AA range:1-50
Specificity:FKRP Polyclonal Antibody detects endogenous levels of FKRP protein.
Formulation:Liquid in PBS containing 50% glycerol, 0.5% BSA and 0.02% sodium azide.
Source:Polyclonal, Rabbit,IgG
Dilution:WB 1:500 - 1:2000. IF 1:200 - 1:1000. ELISA: 1:20000. Not yet tested in other applications.
Purification:The antibody was affinity-purified from rabbit antiserum by affinity-chromatography using epitope-specific immunogen.
Concentration:1 mg/ml
Storage Stability:-15°C to -25°C/1 year(Do not lower than -25°C)
Other Name:FKRP;Fukutin-related protein
Observed Band(KD):50kD
Background: This gene encodes a protein which is targeted to the medial Golgi apparatus and is necessary for posttranslational modification of dystroglycan. Mutations in this gene have been associated with congenital muscular dystrophy, mental retardation, and cerebellar cysts. Several alternatively spliced transcript variants of this gene have been described, but the full-length nature of some of these variants has not been determined. [provided by RefSeq, Oct 2008],
Function:disease:Defects in FKRP are the cause of congenital muscular dystrophy type 1C (MDC1C) [MIM:606612]. Congenital muscular dystrophies (CMD) are a heterogeneous group of autosomal recessive disorders characterized by hypotonia, muscle weakness, and joint contractures that present at birth or during the first 6 months of life and have dystrophic changes on skeletal muscle biopsy. Mental retardation with or without structural CNS changes may accompany some forms. MDC1C is a form of CMD with onset in the first weeks of life and a severe phenotype with inability to walk, muscle hypertrophy, marked elevation of serum creatine kinase, a secondary deficiency of laminin alpha2, and a marked reduction in alpha-dystroglycan expression. Only a subset of MDC1C patients have brain involvements.,disease:Defects in FKRP are the cause of limb-girdle muscular dystrophy type 2I (LGMD2I) [MIM:607155]. LGMD2I
Subcellular Location:Golgi apparatus membrane ; Single-pass type II membrane protein . Secreted . Cell membrane, sarcolemma . Rough endoplasmic reticulum . Cytoplasm . According to some studies the N-terminal hydrophobic domain is cleaved after translocation to the Golgi apparatus and the protein is secreted (PubMed:19900540). Localization at the cell membrane may require the presence of dystroglycan (By similarity). At the Golgi apparatus localizes to the middle-to-trans-cisternae, as assessed by MG160 colocalization. Detected in rough endoplasmic reticulum in myocytes (PubMed:17554798, PubMed:21886772). In general, mutants associated with severe clinical phenotypes are retained within the endoplasmic reticulum (PubMed:15213246). .
Expression:Expressed in the retina (at protein level) (PubMed:29416295). Expressed predominantly in skeletal muscle, placenta, and heart and relatively weakly in brain, lung, liver, kidney, and pancreas (PubMed:11592034).
商品信息已成功复制,启研竭诚为您服务