
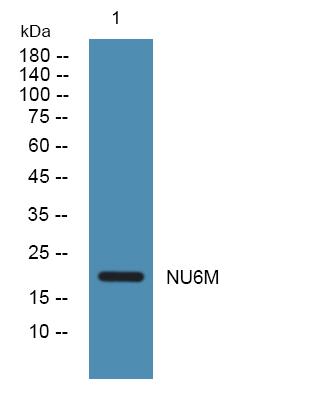
Target:NU6M
Fields:Oxidative phosphorylation;Metabolic pathways;Thermogenesis;Retrograde endocannabinoid signaling;Alzheimer disease;Parkinson disease;Amyotrophic lateral sclerosis;Huntington disease;Prion disease;Pathways of neurodegeneration - multiple diseases;Chemical carcinogenesis - reactive oxygen species;Diabetic cardiomyopathy
Gene Name:MT-ND6 MTND6 NADH6 ND6
Protein Name:NADH-ubiquinone oxidoreductase chain 6 (EC 1.6.5.3) (NADH dehydrogenase subunit 6)
Human Gene Id:4541
Human Swiss Prot No:P03923
Mouse Swiss Prot No:P03925
Rat Swiss Prot No:P03926
Immunogen:Synthesized peptide derived from human protein . at AA range: 30-110
Specificity:NU6M Polyclonal Antibody detects endogenous levels of protein.
Formulation:Liquid in PBS containing 50% glycerol, and 0.02% sodium azide.
Source:Polyclonal, Rabbit,IgG
Dilution:WB 1:500-2000 ELISA 1:5000-20000
Purification:The antibody was affinity-purified from rabbit antiserum by affinity-chromatography using epitope-specific immunogen.
Concentration:1 mg/ml
Storage Stability:-15°C to -25°C/1 year(Do not lower than -25°C)
Observed Band(KD):19kD
Background:catalytic activity:NADH + ubiquinone = NAD(+) + ubiquinol.,disease:Defects in MT-ND6 are a cause of Leber hereditary optic neuropathy (LHON) [MIM:535000]. LHON is a maternally inherited disease resulting in acute or subacute loss of central vision, due to optic nerve dysfunction. Cardiac conduction defects and neurological defects have also been described in some patients. LHON results from primary mitochondrial DNA mutations affecting the respiratory chain complexes.,disease:Defects in MT-ND6 are a cause of Leber hereditary optic neuropathy with dystonia (LDYT) [MIM:500001]; also called familial dystonia with visual failure and striatal lucencies. LDYT is part of a spectrum of Leber hereditary optic neuropathy. It is characterized by the association of optic atrophy and central vision loss with dystonia.,disease:Defects in MT-ND6 are a cause of mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes syndrome (MELAS) [MIM:540000]. MELAS is a genetically heterogenious disorder, characterized by episodic vomiting, seizures, and recurrent cerebral insults resembling strokes and causing hemiparesis, hemianopsia, or cortical blindness.,function:Core subunit of the mitochondrial membrane respiratory chain NADH dehydrogenase (Complex I) that is believed to belong to the minimal assembly required for catalysis. Complex I functions in the transfer of electrons from NADH to the respiratory chain. The immediate electron acceptor for the enzyme is believed to be ubiquinone.,similarity:Belongs to the complex I subunit 6 family.,
Function:catalytic activity:NADH + ubiquinone = NAD(+) + ubiquinol.,disease:Defects in MT-ND6 are a cause of Leber hereditary optic neuropathy (LHON) [MIM:535000]. LHON is a maternally inherited disease resulting in acute or subacute loss of central vision, due to optic nerve dysfunction. Cardiac conduction defects and neurological defects have also been described in some patients. LHON results from primary mitochondrial DNA mutations affecting the respiratory chain complexes.,disease:Defects in MT-ND6 are a cause of Leber hereditary optic neuropathy with dystonia (LDYT) [MIM:500001]; also called familial dystonia with visual failure and striatal lucencies. LDYT is part of a spectrum of Leber hereditary optic neuropathy. It is characterized by the association of optic atrophy and central vision loss with dystonia.,disease:Defects in MT-ND6 are a cause of mitochondrial encephalomyopathy with lacti
Subcellular Location:Mitochondrion inner membrane ; Multi-pass membrane protein .
Expression: Blood,Bone fossil,Bones,Breast cancer,Distant normal tissue,Glioma,
商品信息已成功复制,启研竭诚为您服务




